Webinaire : Présentation de GeVarLi, un outil de santé publique développé pour le projet d’AFROSCREEN
Webinaire : Présentation de GeVarLi, un outil de santé publique développé pour le projet d’AFROSCREEN
Le troisième webinaire organisé dans le cadre du projet AFROSCREEN a été tenu par Nicolas Fernandez, ingénieur en bio-informatique de l’UMI 233 TransVIHMI IRD (Institut de recherche pour le développement – Montpellier). Il a présenté les principes de reproductibilité, de F.A.I.R, d’Open Science et de traçabilité des analyses bio-informatique, appliqués à l’analyse génomique des variants de SARS-CoV-2, à travers le workflow GeVarLi (un pipeline Snakemake pour l’assemblage de génomes), l’appel de variants et l’assignation de lignées pour les SARS-CoV-2 et autres virus émergeants.

Nicolas Fernandez nous a parlé de la crise de la reproductibilité affectant plusieurs domaines scientifiques mais aussi de plusieurs principes et bonnes pratiques permettant de répondre à ce constat. Toute recherche devrait suivre le principe de F.A.I.R., présentant des données : Faciles à trouver (Findable), Accessibles (Accessable), Interopérables (Interoperable) et Réutilisables (Reusable), ainsi qu’à la notion d’Open Science.
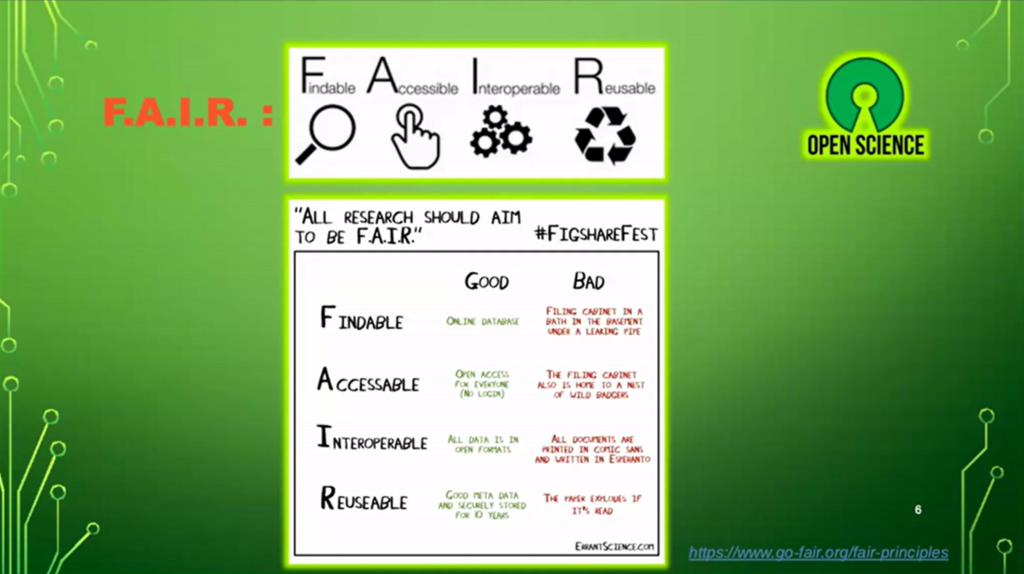
Nicolas Fernandez nous a rappelé l’importance d’appliquer ces principes à nos analyses bio-informatiques, avec l’utilisation de divers outils, comme les gestionnaires de workflows F.A.I.R. (tel que Snakemake, Nextflow), les gestionnaires d’environnements (tel que Anaconda, Docker) et la nécessité de publier et tracer (versionner) son code avec des outils tel que Git (dont l’instance GitLab de l’IRD, IRDForge).
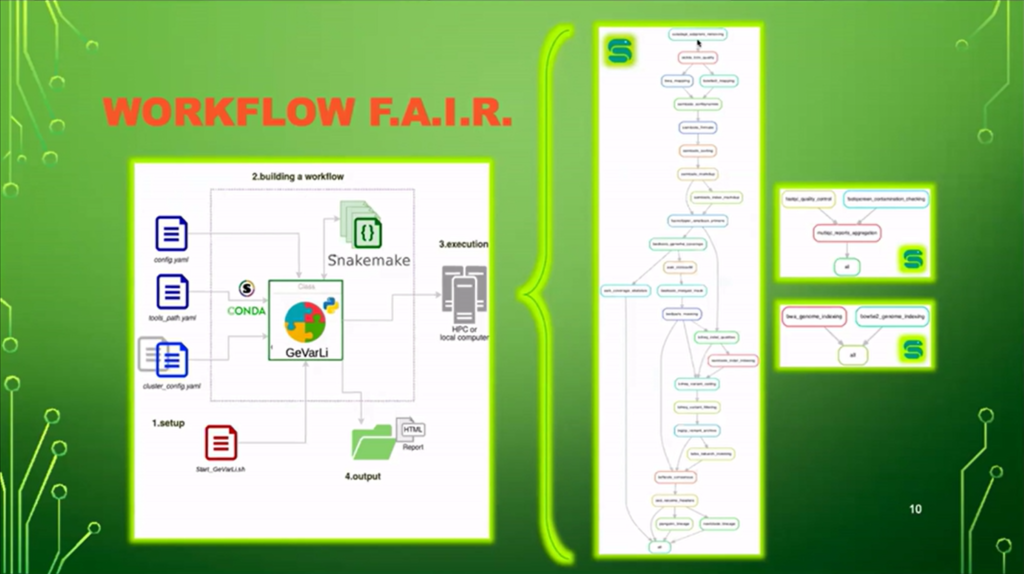
Nicolas Fernandez a ensuite présenté un exemple de workflow F.A.I.R. avec l’outils « GeVarLi : GEnome assembly, VARiant calling and LIneage assignation », un pipeline automatisé en Snakemake développé pour l’analyse génomique de SARS-CoV-2 et autre virus émergeants. Il nous en a présenté sa genèse, où le trouver, comment l’installer et l’utiliser ainsi que les résultats que l’on obtient et ses avantages.

Nicolas Fernandez a insisté en particulier sur le fait que le workflow était ouvert, libre et gratuit, modulable, et ouvert à contribution et participation collaborative, ainsi que l’importance de citer et de remercier les auteurs des outils libres créés par et pour la communauté.
Pour en savoir plus, retrouvez l’intégralité du webinaire :
- FR : https://youtu.be/haBVJdWqNeE
- EN : https://youtu.be/ssEzpN2NkZc
- et plus d’informations sur GeVarli : https://forge.ird.fr/transvihmi/nfernandez/GeVarLi
Nicolas Fernandez nous a parlé de la crise de la reproductibilité affectant plusieurs domaines scientifiques mais aussi de plusieurs principes et bonnes pratiques permettant de répondre à ce constat. Toute recherche devrait suivre le principe de F.A.I.R., présentant des données : Faciles à trouver (Findable), Accessibles (Accessable), Interopérables (Interoperable) et Réutilisables (Reusable), ainsi qu’à la notion d’Open Science.
Nicolas Fernandez nous a rappelé l’importance d’appliquer ces principes à nos analyses bio-informatiques, avec l’utilisation de divers outils, comme les gestionnaires de workflows F.A.I.R. (tel que Snakemake, Nextflow), les gestionnaires d’environnements (tel que Anaconda, Docker) et la nécessité de publier et tracer (versionner) son code avec des outils tel que Git (dont l’instance GitLab de l’IRD, IRDForge).
Nicolas Fernandez a ensuite présenté un exemple de workflow F.A.I.R. avec l’outils « GeVarLi : GEnome assembly, VARiant calling and LIneage assignation », un pipeline automatisé en Snakemake développé pour l’analyse génomique de SARS-CoV-2 et autre virus émergeants. Il nous en a présenté sa genèse, où le trouver, comment l’installer et l’utiliser ainsi que les résultats que l’on obtient et ses avantages.
Nicolas Fernandez a insisté en particulier sur le fait que le workflow était ouvert, libre et gratuit, modulable, et ouvert à contribution et participation collaborative, ainsi que l’importance de citer et de remercier les auteurs des outils libres créés par et pour la communauté.
Pour en savoir plus, retrouvez l’intégralité du webinaire :
- FR : https://youtu.be/haBVJdWqNeE
- EN : https://youtu.be/ssEzpN2NkZc
- et plus d’informations sur GeVarli : https://forge.ird.fr/transvihmi/nfernandez/GeVarLi